Research
|
Click a link for more information about our research interests:
The question(s) The approach Molecular mechanisms regulating chromatin looping |
The question(s)
Proper regulation of gene expression is essential for nearly all biological processes, including the remarkable ability of a single cell to develop into a fully formed organism. And dysregulation of gene expression underlies many diseases. However, understanding gene regulation in mammals comes with a big challenge: Mammalian genomes are enormous. They contain tens of thousands of genes and hundreds of thousands of enhancers. And enhancers, DNA-elements that activate gene expression, can be hundreds of kilobases or even megabases away from the genes that they control. So, how does the cell ensure that the right enhancer contacts the right gene to establish cell-type specific gene expression programs?
|
It is becoming clear that we can only understand mammalian gene regulation if we understand the 3-dimensional folding of the genome, which controls which enhancer talks to which gene promoter. Specifically, architectural proteins - including CTCF and cohesin - fold the genome into spatial domains by forming chromatin loops. By constraining enhancer-promoter contacts, these domains are thought to regulate gene expression. Moreover, genome misfolding through domain disruption can cause cancer by inducing aberrant enhancer-promoter contacts, which results in oncogene activation. Powerful static snapshot approaches such as Hi-C have revealed the existence of these domains and chromatin loops. However, understanding how loops and domains form, persist, dissolve, and function requires an ability to visualize them and dynamically follow them from “birth-to-death”. We develop experimental, super-resolution imaging and computational technologies for visualizing the dynamics of chromatin loops and the key proteins that regulate looping at the single-molecule level in living cells. And we then then apply these tools to address important biological questions:
|
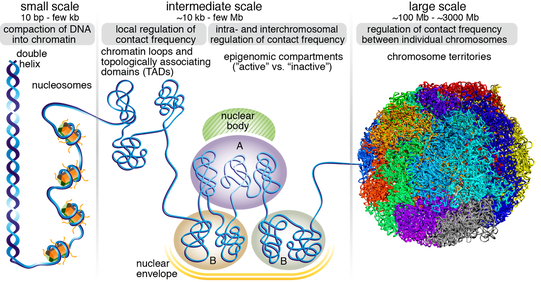
Overview of 3D genome organization at different scales. Figure from Hansen et al. 2018 Nucleus.
|
- What are the molecular mechanisms that regulate chromatin looping?
- How is chromatin looping and transcription regulated during development and dysregulated in disease?
The approach
|
Most biological processes are inherently dynamic and stochastic. Yet, most methods used in biology are time- and/or ensemble-averaged. But to understand a dynamic process, we must use methods that capture its dynamics. For example, if A always occurs just before B, there is a good chance that A is causal for B. Our approach is therefore to develop "birth-to-death" approaches, where we can follow biological processes from beginning to end. That is, we develop new experimental, microscopy and computational methods for tracking these processes at the single-molecule level inside living cells with millisecond and nanometer resolution in time and space (e.g. movie on right). In the case of chromatin looping and enhancer-promoter contact, this means developing new tools that allow us to follow loops inside living cells as they form, persist, function and eventually dissolve.
Following individual molecules inside living cells over time is hard. It requires: 1) new genome-editing and labeling methods; 2) new microscopes for high-resolution imaging; 3) new computational methods for rigorously analyzing and making sense of the data. We therefore take an interdisciplinary approach where biologists, engineers, and physical scientists work closely together to achieve these goals. And we integrate these approaches with traditional methods like biochemistry, genomics and molecular biology in a question-focused manner. |

Tracking a single CTCF protein diffusing and binding inside a live stem cell nucleus to understand how it finds and binds DNA
|
Molecular mechanisms regulating chromatin looping
|
Enhancer-promoter contacts appear to be largely restricted to occur within Topologically Associating Domains (TADs). TADs are formed by the DNA-binding protein CTCF (panel A) and cohesin (panel B). At the molecular level, CTCF binds specific DNA sites and recruits cohesin, which is thought to hold together TADs as a chromatin loop inside its lumen (panel C). Thus, at least two different types of loops shape 3D genome organization: structural loops that form TADs and enhancer-promoter loops that are thought to form inside TADs (panel C). The structural loops are thought to be formed through cohesin-mediated loop extrusion, where cohesin-extrusion is eventually blocked by CTCF (panel D). Consistently, loss of CTCF or cohesin causes loss of most TADs and loops. And functionally, loss of CTCF boundaries can results in human developmental disorders and oncogene activation in (e.g. in glioma) by enabling aberrant enhancer-promoter contacts.
However, we have shown that CTCF and cohesin bind chromatin with very different residence times, suggesting that the complex formed by CTCF and cohesin is unlikely to be a stable complex and thus that TADs and loops are also unlikely to be stable structures inside the cell. Dynamic binding of CTCF and cohesin and the hypothesized mechanism of loop extrusion is nicely illustrated in the video below from the Mirny lab at MIT. |
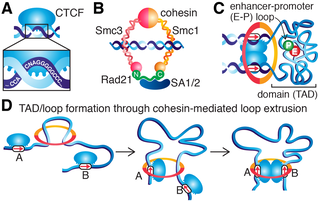
Overview of CTCF, cohesin and loops. Sketches of (A) DNA-binding protein, CTCF, (B) cohesin complex and (C) a CTCF/cohesin-mediated chromatin loop forming a TAD. (D) Simplified sketch of hypothetical loop extrusion mechanism
We are interested in understanding the molecular mechanisms underlying CTCF, cohesin, and loop extrusion and in visualizing loop extrusion in live cells.
How does cohesin extrude loops at speeds exceeding >10-20 kb/min? How and why does CTCF appear to be uniquely able to block cohesin extrusion? Along these lines, we recently discovered key roles for a protein region in CTCF that partially mediates RNA-interactions (RBRi). Loss of the RBRi in mouse embryonic stem cells reduces CTCF clustering and self-interaction, severely affects cell physiology and causes dysregulation of ~500 genes, and leads to the loss of ~1/3 of all chromatin loops. These results suggest that there are different classes of CTCF-mediated chromatin loops. Perhaps this allows the cell to regulate chromatin looping in a cell-type specific manner by regulating binding partners of the RBRi. Above all, it highlights how little we know at the molecular level of how these loops form and are held together and we are interested in elucidating the molecular mechanisms. |
New technologies for tracking single molecules in live cells
The central dogma of molecular biology encompasses 3 major classes of biomolecules: DNA, RNA and proteins. We are developing and improving methods for tracking each class individually and for tracking different classes simultaneously in order to address our motivating questions. For example, for a nuclear DNA-binding protein, a key question is: what fraction of the protein of interest is DNA-bound and what fraction is freely diffusing? And what are the characteristics of these subpopulations? Single-particle tracking (SPT) is ideally suited to answer this, but conventional SPT of nuclear proteins suffer from at least 4 biases: 1) tracking error; 2) motion-blur bias; 3) defocalization bias; 4) analysis bias. We recently developed an integrated approach to combining spaSPT and Spot-On to overcome these biases. spaSPT integrates previous ideas: photo-activation (sptPALM) is used to image at low densities which minimize tracking errors and stroboscopic excitation is used to avoid motion-blurring (the blurring effect observed when imaging a rapidly moving object). To correct for the fact that freely diffusing proteins move out of the focal plane much faster than DNA-bound proteins (movie below on the right show a simulation illutrating this), we extended and validated a prior kinetic modeling framework - now available as Spot-On, a drag-n-drop website interface - for explicitly accounting for defocalization and precisely infer the different subpopulations. For more details, see also the Tools section.
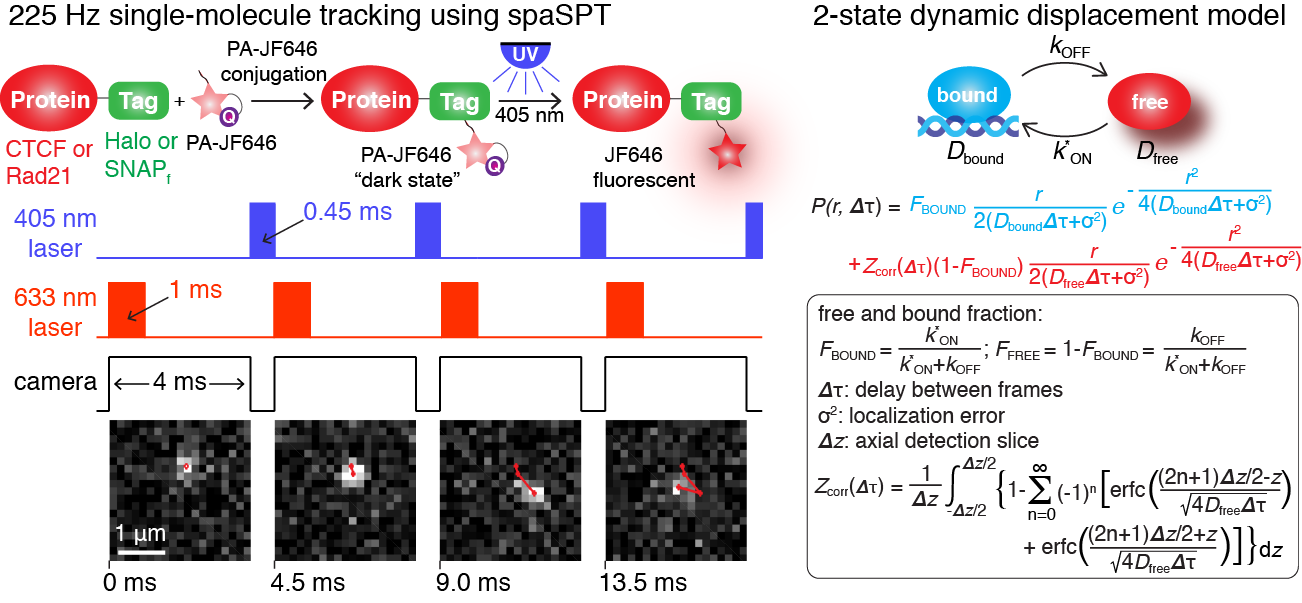
Overview of spaSPT (Left) for tracking single-molecules in live cells with minimal bias and of the kinetic modeling frame-work implemented in Spot-On (Right)
|
|
We are interested in extending these protein SPT approaches to anomalous diffusion as well as developing new experimental and computational approaches for DNA and RNA, and for integrating these different modalities.
Regulation of transcription and looping in development and disease
Our main experimental system is mouse embryonic stem cells. When a stem cell divides, it needs to maintain its transcriptional program: pluripotency genes should be ON and differentiation genes should be OFF. Long-range enhancer-promoter communication plays a key role in the maintenance of such transcriptional programs. But when cells differentiate (e.g. from a stem cell to a neuron), cells need to establish completely new transcriptional programs and long-range enhancer promoter contacts appear to be especially important in these cases as illustrated below.
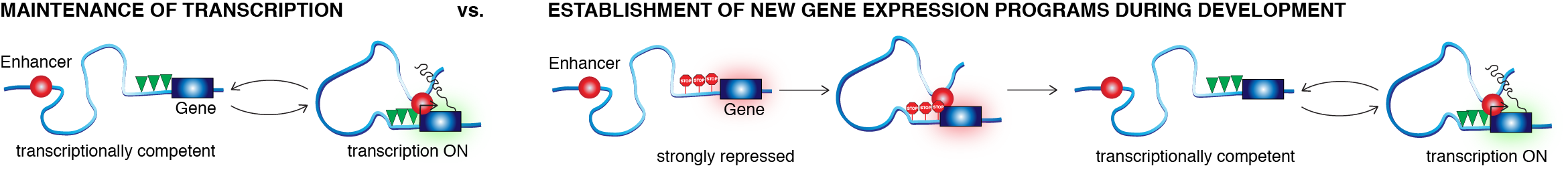
So, how does this work? Questions motivating these efforts include:
For the same reason that enhancer-promoter looping can activate a developmental gene to induce differentiation, aberrant enhancer-promoter looping (e.g. through mutation of CTCF, cohesin or their binding sites) can cause disease. After all, many diseases are caused by expressing the wrong gene at the wrong time. Dysregulation of chromatin looping appears to occur especially frequently in cancer and we are interested in applying our imaging approaches to elucidate the molecular mechanisms through which dysregulation of looping causes cancer.
- How and to what extent does 3D genome organization and looping regulate gene expression during development?
- What are the molecular mechanisms through which cell-type specific regulation is achieved?
- How are the dynamics regulated during development?
For the same reason that enhancer-promoter looping can activate a developmental gene to induce differentiation, aberrant enhancer-promoter looping (e.g. through mutation of CTCF, cohesin or their binding sites) can cause disease. After all, many diseases are caused by expressing the wrong gene at the wrong time. Dysregulation of chromatin looping appears to occur especially frequently in cancer and we are interested in applying our imaging approaches to elucidate the molecular mechanisms through which dysregulation of looping causes cancer.
Computational models of genome organization and transcription
We integrate our experimental results with mechanistic computational models through an iterative "watch, perturb and learn" approach. After all, a mechanistic model is just a mathematical description of our assumptions. Our vision is to take an iterative approach: we apply our imaging technologies for precision measurements of dynamics and mechanisms. We use the measurements to develop and parameterize mechanistic models. To test our understanding, we make model predictions, which we go back and test. Although many model predictions may fail, the manner in which they fail, tells us what which of our mechanistic assumptions were wrong. Thus, by iteratively going through this combined experimental and computional approach, our aim is to eventually derive mechanistically accurate and quantitatively predictive models of genome folding and gene expression.
A synthetic biology approach to 3D genome organization
|
How will we know if we understand genome organization and how it relates to transcription? The ultimate proof that we understand something, is our ability to build it. Therefore, our long-term goal is to reach a predictive and quantitative understanding of 3D genome organization sufficient to allow us to de novo design megabase-sized segments with multiple genes, domains and loops, that fold in predictable ways and exhibit desired gene expression dynamics and cell-type specificities.
If we can achieve this milestone, it suggests that we understand genome organization. But equally importantly, we may be able to correct genome misfolding in disease, design tools to modulate specific loops in a predictable manner and there are countless synthetic biology and therapeutic applications (e.g. gene therapy). We will work towards this ultimate goal initially using simple systems and increase the complexity as our understanding grows. |
Outline of computationally guided de novo design challenge
|
Interested?
Does any of this sound interesting? Or do you have your own ideas related to these directions? We are actively looking for new graduate students, post-docs and research assistants to Join our team. We are an interdisciplinary lab and excited to welcome new members from a range of backgrounds including biology, engineering, and the physical sciences. Please see the Join page for more information.